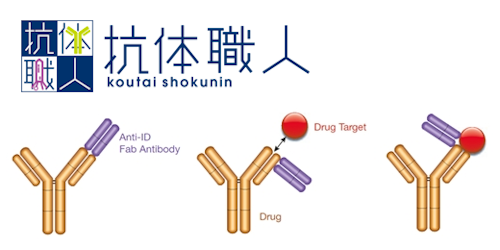
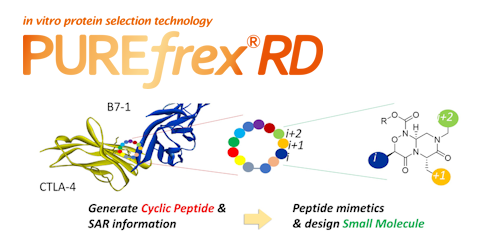
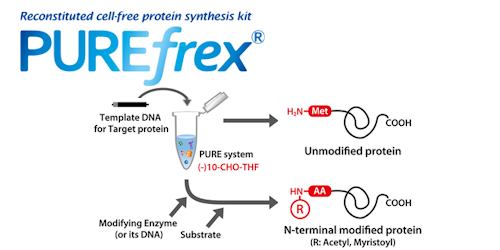
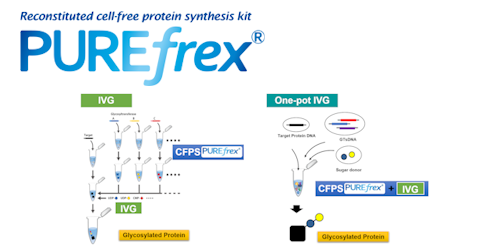
トピックス PUREfrex® 展示会・学会 第25回 日本蛋白質科学会年会で発表いたしました PUREfrex® その他 PUREfrex®を用いたタンパク質の脂質修飾および膜への局在に関する論文が掲載されました 抗体職人 抗イディオタイプ抗体の作製 PUREfrex® 製品・サービス 新製品のお知らせ 【大容量! 50 mL規格】 PUREfrex® その他 PUREfrex®を用いたタンパク質合成におけるN末端アミノ酸配列の影響に関する論文が掲載されました PUREfrex® RD Drug Discovery その他 CTLA-4結合環状ペプチドおよびその低分子模倣化合物に関する論文が掲載されました PUREfrex® その他 PUREfrex®を用いたタンパク質翻訳後修飾に関する論文が掲載されました PUREfrex® 糖タンパク質の合成
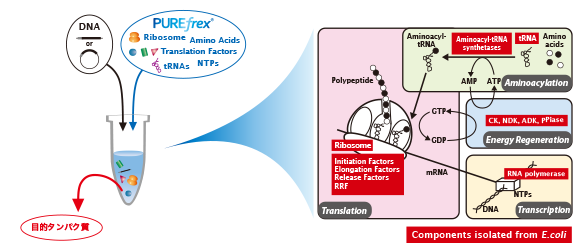
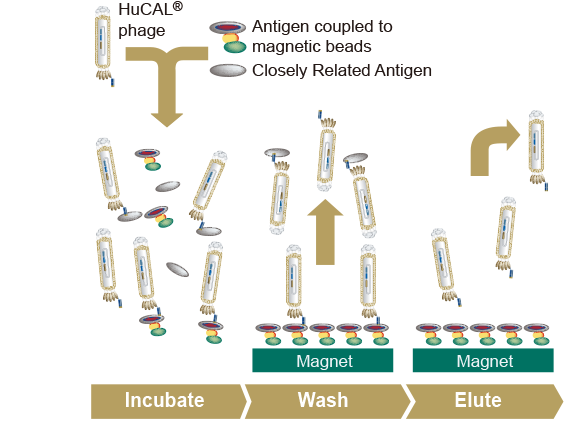
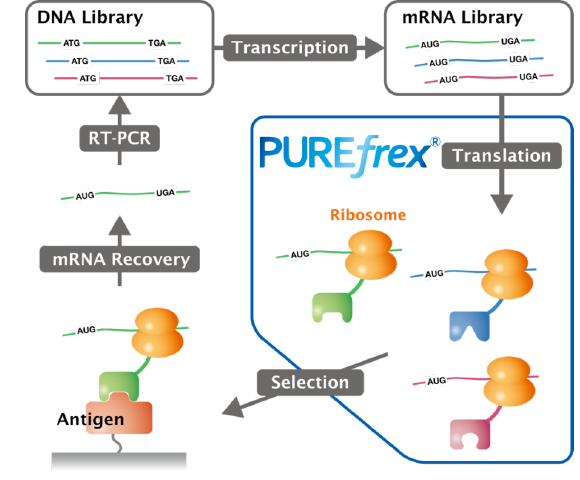
ジーンフロンティアがご提供する製品・サービス PUREfrex® 再構成型無細胞タンパク質合成キット 抗体職人 カスタムモノクローナル抗体作製サービス PUREfrex® RD バイオ医薬開発のためのスクリーニング技術 Drug Discovery ユニークな技術に基づく革新的な創薬シーズ
PUREfrex® 再構成型無細胞タンパク質合成キット TOP 概要 製品一覧 技術情報 ダウンロード 事例紹介 論文 よくある質問 お問い合わせ 詳細を見る 抗体職人 カスタムモノクローナル抗体作製サービス TOP 価格 サービスの流れ ダウンロード 事例紹介 論文 よくある質問 お問い合わせ 詳細を見る PUREfrex® RD バイオ医薬開発のためのスクリーニング技術 TOP ダウンロード 事例紹介 論文 よくある質問 お問い合わせ 詳細を見る Drug Discovery ユニークな技術に基づく革新的な創薬シーズ 概要 お問い合わせ 詳細を見る
ニュース PUREfrex®を用いたタンパク質の脂質修飾および膜への局在に関する論文が掲載されました その他 PUREfrex® 2025.06.26 第25回 日本蛋白質科学会年会で発表いたしました 展示会・学会 PUREfrex® 2025.06.10 システムメンテナンスのお知らせ その他 2025.06.06 第47回 日本分子生物学会年会でポスター発表いたしました 展示会・学会 PUREfrex® 2024.12.09 EdgeブラウザでPDFが正しく表示されない問題について その他 2024.11.28 もっと見る 事例紹介 PUREfrex® N末端修飾タンパク質(アセチル化、ミリストイル化)の合成 2023.06.26 抗体職人 抗イディオタイプ抗体の作製 2023.05.22 PUREfrex® 糖タンパク質の合成 2022.07.08 抗体職人 リン酸化された Ser を見分ける抗体 2022.04.14 抗体職人 1箇所のアミノ酸置換を見分ける抗体の作製例 2022.01.21 もっと見る